

Abstract
BACKGROUND:
Glanzmann's thrombasthenia (GT) is an inherited platelet disorder (IPD) that leads to clinically significant bleeding. Platelets from patients with GT can show qualitative or quantitative defects of platelet membrane glycoprotein (GP) IIb/IIIa complex. Most GT are caused by autosomal recessive genetic defects in ITGA2B and ITGB3 (genes for GPIIb and GPIIIa, respectively) with rare cases showing an autosomal dominant pattern. Accurate diagnosis of GT requires a constellation of both phenotypic and genetic studies. Here we report a unique case of autosomal dominant GT resulted from thorough phenotypic and genetic studies.
PATIENTS AND METHODS:
A 19 year-old woman was recently evaluated for life-long history of easy bruising and severe menorrhagia that was only partially responsive to medroxyprogesterone. She has severe thrombocytopenia first recognized shortly after birth with a platelet count around 20x109/L. Platelet size was normal. She was presumed to have immune-mediated thrombocytopenia (ITP) since early infancy, but lacking responsiveness to the conventional treatments of ITP including immunomodulation, splenectomy and thrombopoietin receptor agonists. Family history was negative for a bleeding diathesis. Both von Willebrand factor antigen and activity were within normal range. Bone marrow aspirate and biopsy with associated chromosome studies were all normal. Platelet surface glycoprotein assessment by flow cytometry using antibodies to GP IIb, IIIa, Ia, Ib-a, VI, IX, and whole exome sequencing (WES) utilizing a predefined list of 53 clinically significant genes* related to genetically IPDs were performed. Due to thrombocytopenia platelet aggregation studies could not be performed.
METHODS/RESULTS:
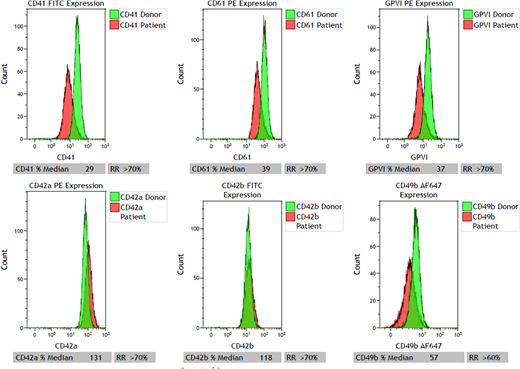
Platelet surface expression of GPIIb (CD41) and GPIIIa (CD61) were markedly decreased suggestive of a variant of GT (Figure). WES performed with Illumina HiSeq 2500 sequencing system by using Agilent SureSelelct CRE kit V1 to capture and target the exonic regions showed presence of a heterozygous mutation in ITGB3 gene (c2213T>G; p.Leu738Arg). By in silico prediction modeling, this mutation is likely to be pathogenic and results in the substitution of hydrophobic leucine with hydrophilic arginine in the transmembrane domain of the β3 subunit of alpha IIb/beta 3 integrin (αIIbβ3), the platelet receptor that binds to fibrinogen. Interestingly, GPVI is also decreased which may be associated with GPIIb and GPIIIa deficiency or due to accelerated shedding since no GPVI mutation was identified.
CONCLUSION:
We describe a patient with GT associated with a novel heterozygous autosomal dominant mutation in ITGB3 gene with substitution of leucine with arginine (c2213T>G; p.Leu738Arg). This deletion caused a low expression of αIIbβ3 integrin on her platelets surface and severe thrombocytopenia. This case underscores the importance of an integrated phenotyping and genotyping approach to render a definitive diagnosis of an IPD.
*Platelet exome gene list: ABCG5, ABCG8, ACBD5, ACTN1, ANKRD26, ANO6, AP3B1, BLOC1S3, BLOC1S6, CYCS, DTNBP1, ETV6, FLI1, FLNA, GATA1, GFI1B, GP1BA, GP1BB, GP6, GP9, HOXA11, HPS1, HPS3, HPS4, HPS5, HPS6, ITGA2B, ITGB3, LYST, MASTL, MPL, MYH9, NBEAL2, ORAI1, PSRX1, P2RY12, PLA2G4A, PRKACG, RASGRP2, RBM8A, RUNX1, STIM1, STX11, STXBP2, TBXA2R, TBXAS1, THPO, TUBB1, UNC13D, VIPAS39, VPS33B, VPS45, WAS
No relevant conflicts of interest to declare.

